Transition metal complexes have been playing an important role in research for cancer treatment since the discovery of cisplatin in the 1960s.1 Among the non-platinum compounds, ruthenium complexes have been studied most extensively and two of them, namely NAMI-A and KP1019/KP1339, have successfully entered clinical trials.2,3 In these octahedral complexes the ruthenium ion is in the oxidation state +3 and studies have shown that stepwise dissociation of the four chlorido ligands and reduction to the oxidation state +2 are essential steps in their mechanism of action.
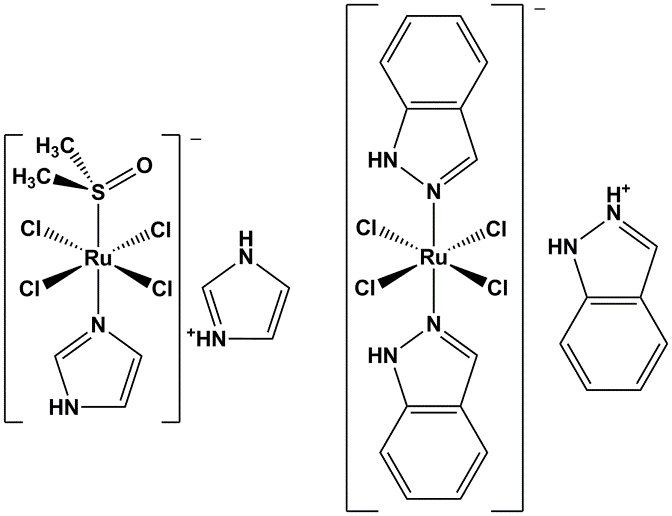
Figure 1: NAMI-A (left) and KP1019 (right), two ruthenium compounds which completed phase II clinical trials.
In the past fifteen years the focus of the research has mainly shifted towards ruthenium(II) complexes. These complexes most often bear a face-capping ligand which stabilizes the oxidation state +2 and allows the fine-tuning of the physico-chemical properties as it reduces the number of labile halide ligands. At first, inspired by the research on Pt drugs, organoruthenium complexes bearing simple amine, halido and carboxylato ligands were studied and several highly cytotoxic ruthenium species were identified.4 However, it quickly became apparent that unlike platinum drugs DNA is not always the main target of organoruthenium compounds. While some complexes do interact with DNA different protein targets recently seem more plausible.4,5
In the past decade the research group of Prof. I. Turel has studied ruthenium complexes with purine antiviral agents,6 neurotransmitter histamine,7 antibacterial agents from the quinolone family8-11 and azole antifungal agents.12 Complexes of quinolones were especially thoroughly studied. The organometallic ruthenium complex of quinolone ofloxacin was found to cause DNA shrinkage and prevent the binding of cisplatin in a competitive binding experiment.19 Similar complexes were also isolated with other quinolone family members and their cytotoxicity against various cancer cell lines and inhibitory potency against two enzymes of the cathepsin family were tested.8,9 Ruthenium(II)-quinolone complexes where 1,4,7-trithiacyclononane replaces the arene as the face-capping ligand were also prepared and interactions with possible targets were studied by UV−Vis spectroscopy, cyclic voltammetry, and viscosity measurements. The complexes strongly bind to serum proteins and intercalate DNA.9 These studies have clearly confirmed that quinolone antibacterial agents may be converted to ruthenium complexes that exert interesting physico-chemical and biological properties. Moreover, fine-tuning of these properties can be achieved by minor changes in the molecular structure of the complexes.
We have recently reported the improved synthesis of an organoruthenium-clioquinol complex (1) which exhibits selective toxicity towards leukaemic cancer cells (Figure 2).13 In our efforts to ‘teach an old dog new tricks’, we combined in a single molecule the structural fragment of an established apoptotic agent, clioquinol, with an organoruthenium moiety. The conjugation of a Ru species to the CqH ligand led to induction of apoptosis at significantly lower concentrations if compared to the CqH alone. Moreover, these mechanisms were copper independent and involved the non-proteasomal inhibition of the NFκB signalling pathway. No effects on the cell-cycle distribution could be observed, which altogether suggests a mechanism of action that is different from that of the free ligand.13
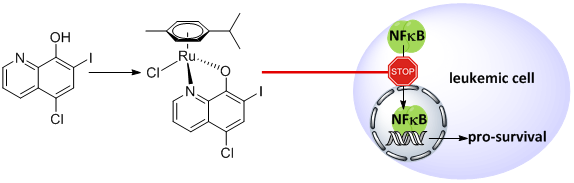 Figure 2: TOC graphic from 13.
Figure 2: TOC graphic from 13.
Very recently we reported a series of organoruthenium(II) chlorido complexes with fluorinated diketonate ligands [(η6–p-cymene)Ru(F3C–acac–Ar)Cl] (Figure 3) and their respective 1,3,5-triaza-7-phosphaadamantane (pta) derivatives [(η6–p-cymene)Ru(F3C–acac–Ar)pta]PF6 (Figure 3).14 All complexes were inactive against non-malignant keratinocytes but displayed variable activity against cancer cell models (ovarian, osteosarcoma). Despite a marginally lower cellular Ru accumulation compared to the chlorido complexes, pta analogues showed higher activity especially in the osteosarcoma model. Reduction of glutathione levels by buthionine-sulfoximine (BSO) significantly enhanced the activity of all compounds with the most pronounced effects being observed for the pta series resulting in IC50 values down to the nanomolar range. While all chlorido complexes potently induce reactive oxygen species, DNA damage, and apoptosis, the respective pta compounds widely lacked ROS production but blocked cell cycle progression in G0/G1 phase.
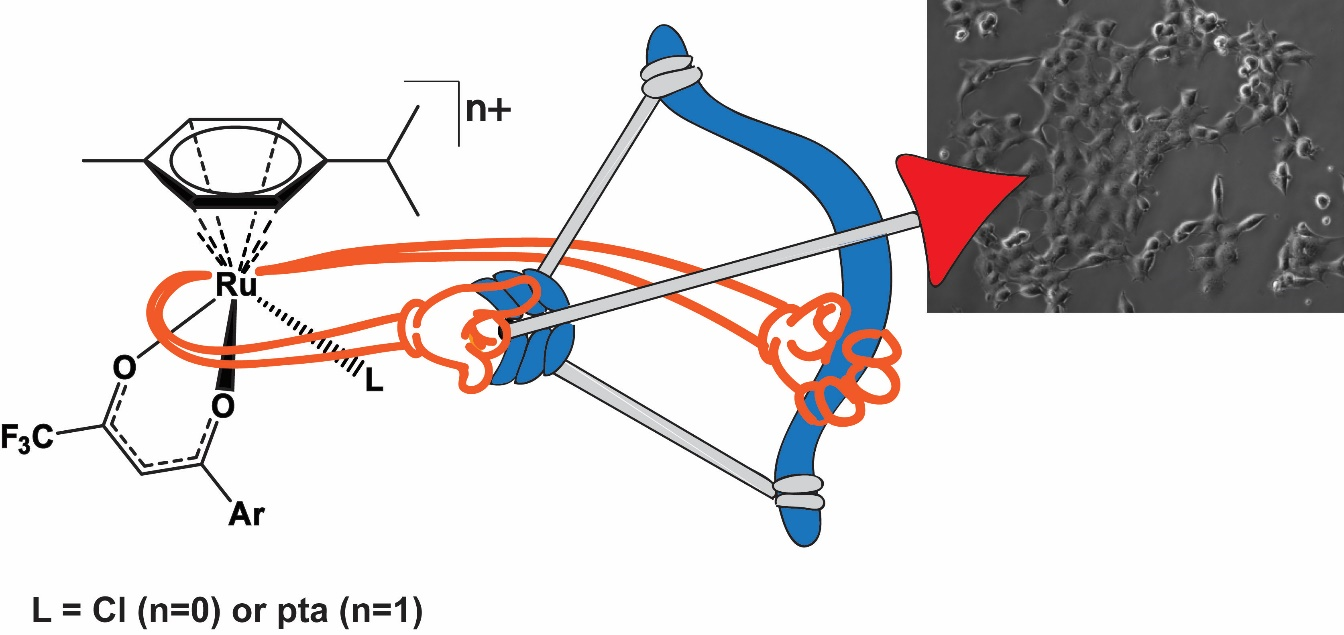 Figure 3: TOC graphic from 14.
Figure 3: TOC graphic from 14.
- Rosenberg, B.; Vancamp, L.; Trosko, J. E.; Mansour, V. H., Nature 1969, 222, 385-386.
- Alessio, E., Bioinorganic Medicinal Chemistry. Wiley-VCH Verlag & Co. KGaA: Weinheim, Germany, 2011.
- Hartinger, C. G.; Zorbas-Seifried, S.; Jakupec, M. A.; Kynast, B.; Zorbas, H.; Keppler, B. K., J. Inorg. Biochem. 2006, 100, 891-904; Sava, G.; Alessio, E.; Bergamo, A.; Mestroni, G., In Topics in biological inorganic chemistry, Clarke, M. J.; Sadler, P. J., Eds. Springer Verlag: Berlin, 1999; pp 143-169; Dyson, P. J.; Sava, G., Dalton Trans. 2006, 16, 1929-1933.
- Habtemariam, A.; Melchart, M.; Fernandez, R.; Parsons, S.; Oswald, I. D. H.; Parkin, A.; Fabbiani, F. P. A.; Davidson, J. E.; Dawson, A.; Aird, R. E.; Jodrell, D. I.; Sadler, P. J., J. Med. Chem. 2006, 49, 6858-6868.
- Kilpin, K. J.; Dyson, P. J., Chem. Sci. 2013, 4, 1410-1419; Hartinger, C. G.; Dyson, P. J., Chem. Soc. Rev. 2009, 38, 391-401; Ang, W. H.; Casini, A.; Sava, G.; Dyson, P. J., J. Organomet. Chem. 2011, 696, 989-998; Sava, G.; Bergamo, A.; Dyson, P. J., Dalton Trans. 2011, 40, 9069-9075.
- Turel, I.; Pecanac, M.; Golobic, A.; Alessio, E.; Serli, B., Eur. J. Inorg. Chem. 2002, 8, 1928-1931.
- Kljun, J.; Petriček, S.; Žigon, D.; Hudej, R.; Miklavčič, D.; Turel, I., Bioinorg. Chem. Appl. 2010, art. no. 183097.
- Hudej, R.; Kljun, J.; Kandioller, W.; Repnik, U.; Turk, B.; Hartinger, C. G.; Keppler, B. K.; Miklavcic, D.; Turel, I., Organometallics 2012, 31, 5867-5874.
- Kljun, J.; Bratsos, I.; Alessio, E.; Psomas, G.; Repnik, U.; Butinar, M.; Turk, B.; Turel, I., Inorg. Chem. 2013, 52, 9039-9052.
- Kljun, J.; Bytzek, A. K.; Kandioller, W.; Bartel, C.; Jakupec, M. A.; Hartinger, C. G.; Keppler, B. K.; Turel, I., Organometallics 2011, 30, 2506-2512.
- Turel, I.; Kljun, J.; Perdih, F.; Morozova, E.; Bakulev, V.; Kasyanenko, N.; Byl, J. A. W.; Osheroff, N., Inorg. Chem. 2010, 49, 10750-10752.
- Kljun, J.; Scott, A. J.; Lanišnik-Rižner, T.; Keiser, J.; Turel, I., Organometallics, 2014, 33, 1594-1601.
- Gobec, M.; Kljun, J.; Sosič, I.; Mlinarič-Raščan, I.; Uršič, M.; Gobec, S.; Turel, I., Dalton Trans. 2014, 43, 9045-9051.
- Seršen, S.; Kljun, J.; Kryeziu, K.; Panchuk, R.; Alte, B.; Körner, Heffeter, P.; Berger, W.; Turel, I., J. Med. Chem. 2015, 58, 3984-3996.